
- Summary
- Overview of 21 CFR Part 11 Compliance in Clinical Trials
- What Is 21 CFR Part 11 Compliance?
- Structure of 21 CFR Part 11: Subparts and System Scope
- Why 21 CFR Part 11 Compliance Was Introduced
- Who Must Comply with 21 CFR Part 11 and Which Systems Are Covered
- Key Requirements for 21 CFR Part 11
- Technical Controls Supporting 21 CFR Part 11 Compliance
- System Validation Lifecycle in 21 CFR Part 11 Compliance
- Common Challenges in 21 CFR Part 11 Compliance Management
- FDA Inspection Readiness for 21 CFR Part 11 Compliance
- Best Practices for Maintaining 21 CFR Part 11 Compliance
- Conclusion
Summary
21 CFR Part 11 compliance defines how electronic records and electronic signatures can be used in FDA-regulated clinical trials and research environments. The regulation outlines requirements for system validation, audit trails, access controls, electronic signatures, and data integrity to help ensure electronic records remain accurate and traceable throughout the clinical trial lifecycle.
Overview of 21 CFR Part 11 Compliance in Clinical Trials
Clinical trials generate large volumes of data, and for many years, these records were managed on paper. But as organizations increasingly rely on electronic systems for clinical research, there is a growing need to ensure that digital records remain accurate and traceable.
This shift led to the introduction of 21 CFR Part 11 by the U.S. Food and Drug Administration, establishing requirements for the use of electronic records and electronic signatures in FDA-regulated environments.
Without proper controls, these systems can create risks related to unauthorized access, missing audit trails, and inconsistent records. 21 CFR Part 11 addresses these concerns through requirements for system validation, audit trails, electronic signatures, access controls, and data integrity.
What Is 21 CFR Part 11 Compliance?
21 CFR Part 11 is a regulation issued by the U.S. Food and Drug Administration that defines how electronic records and electronic signatures can be used in place of paper records and handwritten signatures in regulated environments.
The regulation applies to electronic records that are created, modified, maintained, archived, retrieved, or transmitted under FDA regulations. Its primary objective is to ensure that electronic records remain trustworthy and accurate throughout their lifecycle.
If an FDA predicate rule requires a record to be maintained, then the electronic version of that record typically falls under Part 11 requirements.
For example, Part 11 may apply to:
electronic case report forms (eCRFs)
informed consent records
laboratory data
trial management documentation
electronic signatures used for approvals or reviews
The regulation also establishes requirements for electronic signatures, ensuring that they are uniquely linked to individuals and cannot be reused or reassigned improperly.
Rather than focusing only on software functionality, 21 CFR Part 11 emphasizes accountability and data integrity across electronic records and signatures.
Structure of 21 CFR Part 11: Subparts and System Scope
21 CFR Part 11 is divided into multiple sections that define requirements for electronic records and electronic signatures used in FDA-regulated environments.
The regulation is generally organized into three primary subparts.
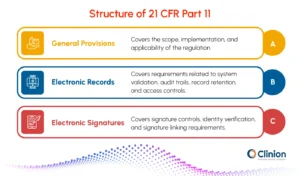
Subpart A - General Provisions:
Defines the scope of 21 CFR Part 11 and explains when the regulation applies to electronic records and electronic signatures used in FDA-regulated environments.
Subpart B - Electronic Records:
Establishes requirements for managing electronic records, including system validation, audit trails, record retention, and controlled system access.
Subpart C - Electronic Signatures:
Defines the requirements for electronic signatures, including user identity verification, signature controls, and permanent linking of signatures to electronic records.
The regulation also distinguishes between closed systems and open systems.
A closed system is an environment in which access to the system is controlled by the organization responsible for the records. Examples may include internally managed EDC platforms or restricted clinical databases used within a sponsor or CRO environment.
An open system involves environments where records may pass through networks or systems outside direct organizational control. Because these environments may introduce additional security risks, organizations often implement extra safeguards such as encryption, secure transmission protocols, and stronger authentication controls.
Understanding these sections helps organizations determine which controls apply to their systems, electronic records, and operational workflows.
Why 21 CFR Part 11 Compliance Was Introduced
As clinical trials adopted electronic systems, organizations began replacing paper-based workflows with digital platforms. This transition improved accessibility and operational efficiency, but it also introduced new risks. Electronic systems made it possible to modify records without visible evidence, overwrite historical data, or access sensitive information without proper authorization.
This shift created the need for a regulatory framework that ensures electronic records remain reliable during inspection and oversight activities.
21 CFR Part 11 was introduced to address concerns such as:
- unauthorized changes to records
- incomplete audit histories
- lack of user accountability
- weak security controls
Discover the key benefits in Why Implement an FDA 21 CFR Part 11 Software System
Who Must Comply with 21 CFR Part 11 and Which Systems Are Covered
21 CFR Part 11 applies to regulated entities involved in FDA-regulated activities that use or manage electronic records.
These organizations commonly include:
- Pharmaceutical Companies
- Biotechnology Companies
- Medical Device Organizations
- Sponsors
- CROs
- Clinical Investigators
- IRBs
- Laboratories
The regulation also applies to computerized systems that manage regulated records.
System Type | Example Use in Clinical Trials | Why Part 11 Applies |
Clinical data collection | Stores regulated subject data | |
Trial documentation management | Maintains essential trial documents | |
Electronic informed consent | Captures participant signatures | |
Randomization and drug supply | Supports regulated trial workflows | |
Trial operations tracking | Manages operational trial records | |
Laboratory Systems | Sample and result management | Handles regulated laboratory data |
Although software vendors often build systems with compliance-supporting features, responsibility for compliance ultimately remains with the regulated organization using the system.
For example, a sponsor using a third-party EDC platform is still responsible for ensuring that the system is validated and properly controlled.
Key Requirements for 21 CFR Part 11
21 CFR Part 11 establishes both technical and procedural controls for electronic records and electronic signatures.
The regulation focuses on several major areas.
Requirement | What It Means | Why It Matters |
System Validation | Systems should perform consistently as intended | Helps maintain reliable records |
Audit Trails | Tracks changes made to records | Supports traceability |
Access Controls | Limits system access to authorized users | Prevents unauthorized activity |
Electronic Signatures | Links signatures to individual users | Supports accountability |
Record Retention | Preserves records throughout retention periods | Ensures inspection readiness |
System Validation Requirements
One of the most important requirements under Part 11 is system validation.
Validation demonstrates that a computerized system consistently performs according to its intended use. Simply implementing software is not enough. Organizations must document that the system performs as intended and supports data integrity across regulated workflows. Validation activities commonly include:
- defining system requirements
- testing functionality
- documenting expected outcomes
- verifying security controls
- maintaining change control procedures
A risk-based validation approach is commonly recommended. Systems or functions that directly impact patient safety, product quality, or data integrity typically require more extensive testing and documentation.
Audit Trails and Change Tracking
Audit trails are a central component of Part 11 compliance. An audit trail records:
- who made a change
- what was changed
- when the change occurred
These records must be computer-generated and protected from unauthorized modification.
For example, if a user updates laboratory values in an EDC system, the audit trail should preserve:
- the original value
- the updated value
- the timestamp
- the user responsible for the modification
Organizations should not disable or overwrite audit trails. During inspections, regulators often review audit logs to verify data integrity and traceability.
Electronic Signatures Under Part 11
Electronic signatures used in regulated systems must be uniquely linked to individual users.
Part 11 requires organizations to ensure that:
- signatures cannot be reassigned
- users are properly authenticated
- signature records remain linked to associated documents
- signed records include the signer’s name, date, and meaning of the signature
Each electronic signature must display the signer’s identity, timestamp, and the meaning of the action performed, such as approval or review. These details are commonly referred to as signature manifestations and must remain permanently linked to the associated electronic record.
For instance, when a clinical investigator signs off on study data, the system should clearly document:
- who signed
- when the signature occurred
- what action was approved
Shared logins or group credentials create accountability concerns and are considered non-compliant practices.
Access Controls and User Authentication
Part 11 requires organizations to restrict system access to authorized individuals.
This often involves:
- unique user IDs
- password policies
- role-based permissions
- multi-factor authentication
- account lockout procedures
- prompt deactivation of inactive accounts
Access controls help prevent unauthorized users from viewing, modifying, or approving regulated records.
For example, a clinical coordinator may have permission to enter data into an EDC system, while only investigators or designated reviewers can approve records electronically.
Data Integrity and ALCOA+ Principles
Part 11 compliance is closely connected to data integrity principles. Organizations commonly use the ALCOA+ framework to evaluate the quality and reliability of regulated data.
ALCOA+ refers to records being:
- Attributable
- Legible
- Contemporaneous
- Original
- Accurate
- Complete
- Consistent
- Enduring
- Available
These principles help ensure that records remain trustworthy throughout the clinical trial lifecycle.
For example, if source data is modified, the system should preserve the original entry, identify the individual making the change, and record the timing of the update. Without these controls, it becomes difficult to confirm the reliability of clinical data during inspections or submissions.
Technical Controls Supporting 21 CFR Part 11 Compliance
Organizations use several technical safeguards to support compliance with Part 11 requirements. These controls help protect electronic records from unauthorized access and untraceable modifications.
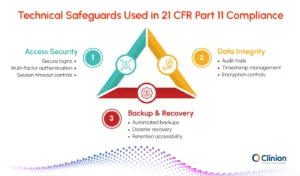
Common technical controls include:
- encryption for data transmission and storage
- secure login procedures
- multi-factor authentication
- centralized timestamp management
- automated backups
- disaster recovery procedures
- audit trail logging
- session timeout controls
Systems used in open environments, particularly cloud-based platforms accessed over public networks, often implement additional encryption and authentication safeguards.
Backup and recovery processes are also important. Electronic records must remain accessible for the full retention period, including during system disruptions or recovery scenarios.
Many organizations periodically test restoration procedures to verify that archived records can be recovered successfully.
System Validation Lifecycle in 21 CFR Part 11 Compliance
System validation is a core requirement under 21 CFR Part 11, ensuring computerized systems operate as intended and maintain control over regulated data.
Since validation involves a structured, multi-stage process, it is explained in more detail separately to provide a clearer understanding of how compliance is achieved in practice.
Validation typically follows a structured lifecycle approach, although the exact process may vary depending on system complexity and regulatory expectations.
Validation Stage | Purpose |
User Requirements Specification (URS) | Defines what the system is expected to do based on business and regulatory needs |
Design Qualification (DQ) | Confirms that the selected system design can support the defined requirements |
Installation Qualification | Verifies that the system and supporting infrastructure are installed correctly |
Operational Qualification (OQ) | Tests whether key system functions operate as expected under controlled conditions |
Performance Qualification (PQ) | Confirms that the system performs effectively in real-world operational use |
Each stage plays a different role in establishing system reliability and compliance.
For example, during Operational Qualification (OQ), organizations test critical functions such as:
- login and password controls
- audit trail generation
- electronic signature workflows
- role-based permissions
- backup and recovery functions
Performance Qualification (PQ) focuses on how the system performs during actual business operations. Users may enter clinical trial data, resolve queries, or complete approval workflows to verify that the system supports intended processes consistently.
Validation is not treated as a one-time activity. Software updates, configuration changes, infrastructure modifications, or new integrations may require additional testing and documentation to maintain the validated state of the system.
Common Challenges in 21 CFR Part 11 Compliance Management
Maintaining Part 11 compliance can become difficult when organizations manage multiple systems and vendors across global trials, as each introduces its own operational challenges.
Some of the most common compliance challenges include:
Common Issue | Potential Risk |
Shared user accounts | Lack of accountability |
Disabled audit trails | Missing change history |
Incomplete validation | Unverified system performance |
Weak password controls | Unauthorized access |
Poor backup procedures | Risk of data loss |
Organizations also face challenges related to:
- maintaining consistent SOPs
- validating legacy systems
- managing spreadsheet-based processes
- coordinating vendor oversight
- ensuring regular staff training
Legacy systems can present particular difficulties when older platforms lack modern audit trail functionality or configurable security controls. Another common issue involves unmanaged spreadsheets used for regulated calculations or data tracking. If these tools are not validated appropriately, they may create compliance risks.
FDA Inspection Readiness for 21 CFR Part 11 Compliance
FDA inspections focus on whether electronic systems maintain a reliable record history and controlled data access throughout their use in clinical trials.
Inspectors may review:
- validation documentation
- audit trails
- access control logs
- SOPs
- training records
- backup and recovery procedures
- system inventories
Organizations are generally expected to demonstrate that electronic records:
- are attributable to specific individuals
- remain accurate and complete
- can be retrieved throughout retention periods
- preserve historical changes
Inspectors may also verify whether audit trails are active and whether organizations review them regularly.
Training records are another important area. Personnel using regulated systems should understand both operational procedures and their responsibilities related to electronic records and signatures.
Inspection readiness often depends on maintaining organized documentation and consistent operational practices rather than preparing only when an audit occurs.
Best Practices for Maintaining 21 CFR Part 11 Compliance
Maintaining compliance requires ongoing oversight rather than one-time implementation activities. Organizations commonly use the following practices to strengthen compliance programs.

Validation:
Document IQ, OQ, and PQ activities to maintain the validated state of regulated systems.
Security:
Enforce unique user credentials and controlled access to regulated electronic records.
Audit Trails:
Enable audit trail logging and review records regularly to support traceability.
SOPs:
Maintain updated procedures governing electronic records, system usage, and compliance workflows.
Training:
Train users on operational procedures and responsibilities related to electronic records and electronic signatures.
Backup & Recovery:
Periodically test restoration procedures to ensure regulated records remain accessible during recovery scenarios.
Additional best practices include:
- maintaining a complete system inventory
- reviewing vendor compliance documentation
- restricting administrative privileges
- documenting configuration changes
- conducting periodic internal audits
- monitoring inactive accounts
- testing disaster recovery procedures regularly
A risk-based approach is commonly recommended. Systems handling critical clinical data or supporting patient safety generally require greater oversight and documentation than lower-risk operational tools.
Conclusion
21 CFR Part 11 establishes how electronic records and electronic signatures are expected to function in regulated clinical environments. Its requirements are reflected in how systems are designed and configured across study operations.
As clinical workflows continue to rely on interconnected digital platforms, compliance depends on how consistently these systems preserve record history, enforce controlled access, and support reviewability of data over time.
In practice, inspection outcomes often reflect not just system capability, but how well those controls are maintained throughout the study lifecycle.
How Clinion Supports 21 CFR Part 11 Compliance
Clinion provides clinical trial platforms designed to support 21 CFR Part 11 compliance requirements across regulated research workflows. Clinion systems include capabilities such as audit trails, electronic signatures, role-based access controls, and validation-ready configurations to support electronic record management in FDA-regulated environments. In addition to 21 CFR Part 11, Clinion platforms are also designed to align with other global regulatory and security standards commonly followed in clinical research.


